Our Terms & Conditions | Our Privacy Policy
Evolution of novel mimicry polymorphisms through Haldane’s sieve and rare recombination
Nearly a hundred years ago, JBS Haldane—one of the architects of Modern Synthesis— predicted that novel adaptive mutations under strong selection will rise rapidly to a high frequency if they are genetically dominant over wild-type1. This is because the new, even partially dominant mutations are exposed to selection instantly, rather than suffer potential loss by random genetic drift, even when they initially appear as heterozygotes1. This is now known as Haldane’s sieve. In undefended prey species or Batesian mimics, the mutation that confers phenotypic similarity to well-protected aposematic models is under strong selection because it confers high fitness by reducing predation pressure. Thus, this adaptive mutation will spread rapidly under strong selection if it is dominant, as Haldane predicted. Indeed, mimetic forms in polymorphic mimicry in Papilio swallowtails are often genetically dominant over non-mimetic forms2. Two critical predictions follow from this: (a) mimetic forms that evolve in succession are likely to be sequentially dominant, and (b) the mimicry gene will show signatures of selective sweeps and/or episodic selection because of a few successive mutations that improve mimetic resemblance. However, testing the Haldane’s sieve hypothesis has been difficult in wild populations since the precise evolutionary chronology of mimetic forms in relation to non-mimetic relatives among these species, the dominance relationships among different female forms, and the precise molecular identities of the underlying genes and alleles have been poorly characterised. Likewise, strongly advantageous novel mutations that sweep through populations may completely replace wild-type alleles, making it difficult to estimate their evolutionary origins and the dominance relationships amongst them. On the contrary, selection for balanced polymorphisms in Batesian mimicry may maintain successive beneficial alleles, preserving their evolutionary histories in relation to the original wild-type (f. cyrus in case of the polytes species group; Fig. 1). This makes mimicry polymorphisms an excellent system to test the predictions of Haldane’s sieve, which we do here by uncovering the missing pieces mentioned above. Finally, we ask how molecular evolution and dominance interact with other genetic aspects to facilitate the evolution of novel forms in an existing polymorphism.
Figure 1:
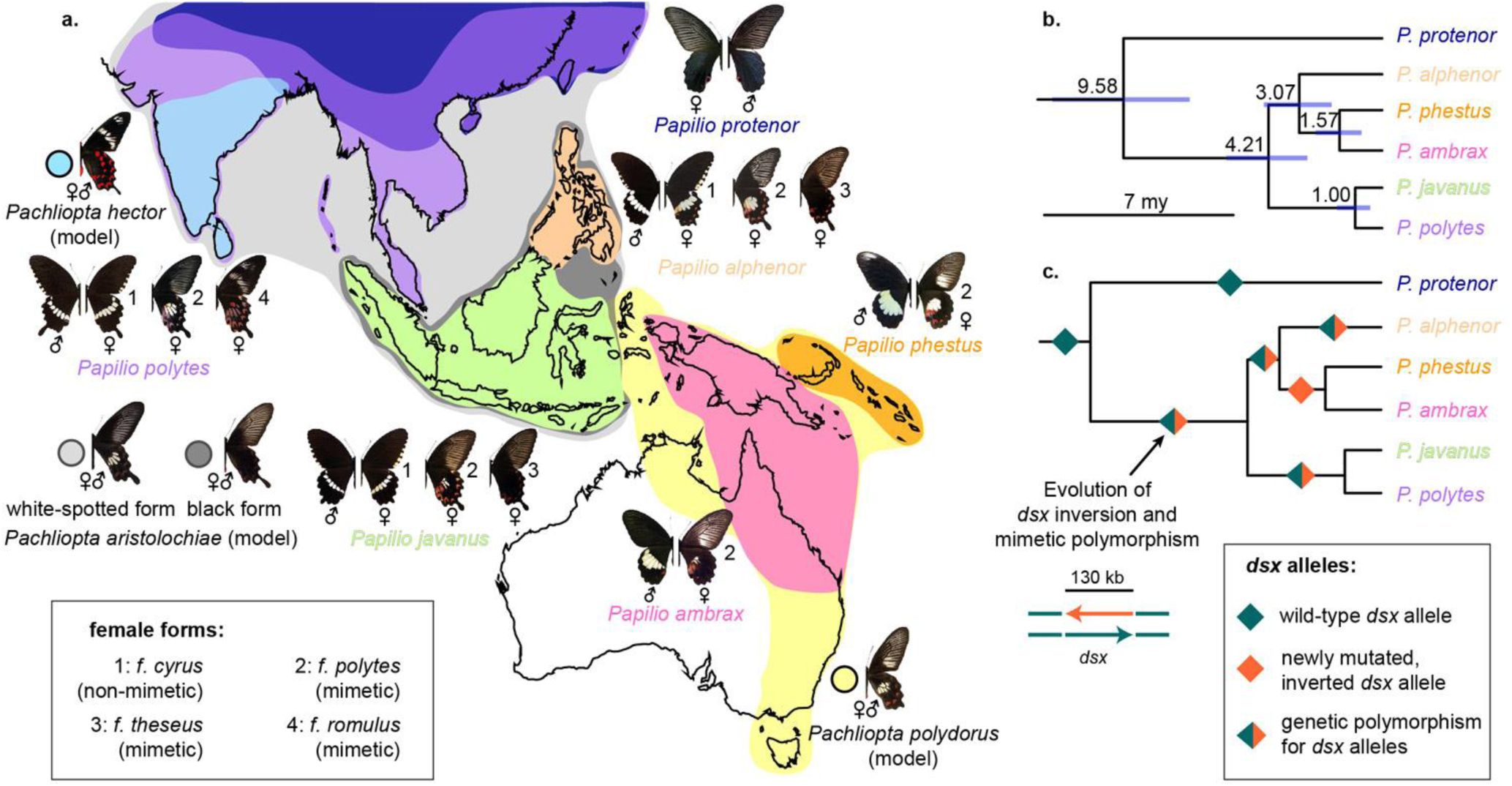
Speciation, wing pattern evolution and mimetic polymorphism in the Papilio polytes species group.
a. Species distributional ranges and wing colour pattern polymorphism in the female forms of the polytes species group, along with their Batesian models. Although the minute details of mimetic wing colour patterns and the presence/absence of tails vary across species and populations, the form names are generalised for the purpose of this paper and apply to multiple species. b. A secondary fossil-calibrated, dated phylogeny of the polytes species group, showing mean with 95% Highest Posterior Density of each split in million years. c. Evolution of mimetic polymorphism and doublesex inversion in relation to speciation events. Diamonds on branches show fixation or polymorphism of female wing patterns and the evolution of accompanying dsx inversion.
Papilio swallowtail butterflies exhibiting polymorphic and female-limited mimicry are classic examples of complex polymorphisms3–5. As edible Batesian mimics, they derive protection from predators by mimicking toxic, warningly patterned (i.e., aposematic) butterflies. Exemplifying this is the Papilio polytes species group, which exhibits four female-limited mimetic morphs or forms (‘f.’): f. cyrus is male-like and non-mimetic, whereas f. polytes, f. theseus and f. romulus are Batesian mimics of aposematic Pachliopta models3,6. Female forms cyrus and polytes occur in several species across the range of the group (Fig. 1a). However, f. romulus is endemic to S. Asia and f. theseus is endemic to islands of SE Asia, because their aposematic models (Pachliopta hector and the black form of Pachliopta aristolochiae, respectively) are endemic to those regions6 (Fig. 1a). The diversity of these mimetic polymorphisms peak in Sri Lanka and India, where forms cyrus, polytes and romulus co-occur7, and in SE Asia where forms cyrus, polytes and theseus co-occur6 (Fig. 1a). The female forms in P. polytes are autosomally inherited and governed by a single locus, H 6,8, which was recently identified to be doublesex (dsx)9,10—a developmental master regulator. Although dsx controls early developmental sex differentiation in insects, it has been co-opted repeatedly in pupal stages to produce ecologically relevant polymorphisms such as polymorphic mimicry in P. polytes, evolving rapidly under positive and pervasive selection11. An inversion spanning the entire mimetic H allele generates a supergene-like architecture and largely prevents recombination 9. How does a single conserved gene such as dsx with a constraining genetic architecture promote the remarkable phenotypic diversity observed across lineages in Papilio?
1. Evolutionary chronology of mimetic forms
We showed recently that P. polytes is not a single species, as traditionally believed6,12, but a group of three allopatric species in two clades: (a) P. polytes (Asian mainland) and its sister P. javanus (mainly Sunda Islands), and (ii) P. alphenor (the Philippines), sister to the species pair P. phestus and P. ambrax (Fig. 1a–b)13. They show hallmarks of distinct species such as well-supported phylogenetic structure, genome-level divergence, strong assortative mate preference, and postzygotic barriers to hybridization13. This discovery reveals the evolutionary history of mimetic polymorphism in this iconic species group, based on the unequal distribution of various female forms across species and populations (Fig. 1). To determine the evolutionary chronology of different female forms along with the underlying mimicry gene inversion among species, we used secondary fossil calibration as previously applied in Papilio14,15 to date the species phylogeny (Fig. 1b–c, Fig. S1; sample details in Table S1). We estimated that the group is 9.58 million years (my) old (95% highest posterior density (HPD): 12.04 to 7.08 my), with the sexually monomorphic P. protenor being basal (Fig. 1b–c, Fig. S2). We used a whole-genome dataset of the polytes group as well as inversion breakpoint-specific primers (Table S2) to track the evolutionary history of the dsx inversion. The orientation of the dsx homologs of outgroups9,16 and the basal P. protenor12 was inferred to be ancestral. Mimicry-related sexual dimorphism, and the dsx inversion, is a derived state shared by all descendent species, and is estimated to have evolved between 9.58 my and 4.21 mya (95% HPD: 5.76 to 2.77 my; Fig. 1b–c, Fig. S2). The ancestral orientation of dsx and f. cyrus were lost, and the dsx inversion and mimetic f. polytes were fixed, in the ancestor of P. phestus and P. ambrax (Fig. 1c). However, both mimicry and inversion polymorphisms were retained in P. alphenor, P. javanus and P. polytes (Fig. 1a, c). Females of these three species share the non-mimetic f. cyrus and mimetic forms f. polytes and f. theseus. We included multiple Batesian models of mimetic female forms that occur over their ranges, including all three models from the Indian subcontinent (Pachliopta aristolochiae, Pachliopta hector and Pachliopta pandiyana) in the time-calibrated phylogenetic reconstruction. The split between Pachliopta aristolochiae and Pachliopta hector is estimated to have occurred around 0.45 mya (95% HPD: 0.82 to 0.15 my; Fig. S2). The wing pattern of Pachliopta aristolochiae is shared among several aposematic Pachliopta and related genera that occupy the mainland and islands in the Indo-Australian region, whereas P. hector has a unique wing pattern with two white bands on the forewing and no white spots on the hindwing (Fig. 1a). Thus, based both on the occurrence of female f. romulus in P. polytes alone (split from its sister P. javanus at approx. 1 my) and the evolution of the unique wing pattern of the Batesian model Pachliopta hector (split from its sister Pachliopta aristolochiae at approx. 0.45 my), we infer the evolution of f. romulus to be less than 0.45 my (95% HPD: 0.82 to 0.15 my) under a new protective umbrella of the S. Asia-endemic Pachliopta hector (Fig. 1b–c, Fig. S2). We tried to find sequences of the dsx inversion breakpoints in the genome sequences of all species in the subgenus Menelaides but found no sequence matches in any species outside of the P. polytes species group. Thus, the mimicry-related inversion found in the P. polytes species group appears to be restricted to that group alone. This implies that whatever long-term balancing selection that different dsx alleles underlying different female forms of P. polytes have experienced also span only the estimated approx. 9.58 my for f. cyrus and f. polytes, and approx. 1 to 0.45 my or shorter for f. romulus. However, smaller inversions in parts of dsx appear to be associated with mimicry in species of the memnon and gambrisius species groups17.
2. Mimetic polymorphism in P. polytes maps to dsx alleles and their expression
The genetic basis of forms cyrus and polytes was earlier mapped to dsx alleles9,10. It was possible that f. romulus of P. polytes is regulated by another gene since it involves pigmentation (red spotting and white bands) that is different from f. polytes (white spotting). We identified the mimicry locus in f. romulus with a genome-wide association study using a romulus-cyrus segregating brood. GWAS revealed 20 sites significantly associated with f. romulus, all of which mapped to the dsx and neighbouring scaffolds that retain synteny with the dsx-containing chromosome 25 in Bombyx mori (Fig. S3, Table S3). Surprisingly, there were no significant hits within the dsx gene, which indicated that reads from f. romulus did not map to the cyrus allele, perhaps due to an inversion and subsequent genetic divergence. To test this possibility, we sequenced whole genomes of 41 wild-caught P. polytes including all three female forms across its geographical range (Fig. S1, Table S1. We found that reads from romulus dsx mapped to the mimetic H allele of f. polytes, and a few reads sparsely mapped to exons of the non-mimetic h allele. This indicated that romulus dsx occurred in the same inverted orientation as the H allele of f. polytes, which we now distinguish as dsx alleles HR and HP, respectively, and h referring to the recessive cyrus allele. To test whether HR and HP have the same dsx inversion, we inspected their breakpoints using breakpoint-specific primers (Table S2) in wild-caught as well as lab-bred individuals, including mapping broods. We found that the breakpoints of HR and HPwere indistinguishable, confirming that the two mimetic alleles have a common evolutionary origin (Fig. S4).
Apart from sharing inversion breakpoints, HR and HP alleles were expressed at similar levels in the developing wings of 3-day old female pupae (mimetic wings) compared to those in males (non-mimetic wings) (Fig. S5). This 3-day pupal stage is a critical window for wing colour pattern differentiation of mimetic females in this species group9,10. However, isoform expression in the female forms was distinct: of the three known female isoforms of dsx, dsxF1 was expressed predominantly in f. romulus wings compared to f. polytes wings (Fig. S5). Moreover, each isoform differs in sequence and structure at the C-terminal end, which potentially alters the DNA-binding ability and facilitates regulation of different downstream effectors by dsx to produce the mimetic and non-mimetic wing patterns10,18. Thus, evidence from genotype-phenotype mapping (Fig. S3), developmental expression (Fig. S5) and allelic characterisation (see below) shows implies that the HR allele and its isoforms genetically and developmentally define and developmentally regulate f. romulus, even though it shares the same inversion breakpoints, many of the defining SNPs and pupal expression levels of the HP allele of mimetic f. polytes.
Similar to f. romulus, the inversion breakpoints of f. theseus were identical to that of f. polytes, as seen in the alignment of genomic data (Fig. S4). This confirmed that the dsx inversion occurred only once and was shared across all the mimetic female forms. However, the dsx coding sequence of f. theseus was identical to that of f. polytes in both P. javanus and P. alphenor (Fig. 3). This suggests that f. theseus may be a developmentally regulated variant of f. polytes in which white spots on hindwings are not expressed. This might also explain why f. polytes and f. theseus show equal dominance6. Similar to f. polytes, f. theseus possibly evolved in the common ancestor of P. javanus and P. alphenor since it is shared between them (Fig. 1a–c). In a previous study, Zhang et. al., (2017) discovered different non-overlapping sets of theseus-specific SNPs in Papilio polytes and Papilio alphenor. Based on our finding that Papilio polytes is a complex of three species, we note that the Zhang et. al. study compared theseus females from P. javanus (sampled from Indonesia) with polytes females from P. polytes (sampled from Japan). Therefore, what they obtained were species-specific differences and not form-specific differences, which were non-overlapping between the two sets of theseus samples. The lack of sequence-level differences between forms polytes and theseus is encapsulated in our gene tree (Fig. S7), where both forms co-occur on the same branch, whether coding or non-coding dsx sequences are considered. Mutations in regulatory elements of dsx or other genes involved in wing patterning and pigmentation pathways affecting developmental regulation alone – which do not affect dominance relationships between forms6 – may explain the maintenance of polytes and theseus forms in the island species. This mechanistic basis regarding its origin needs to be confirmed with further developmental manipulations.
3. Successive mimetic forms are sequentially dominant
An intriguing difference in the genetic basis of wing patterning in Papilio compared to Heliconius butterflies is the nature of genetic dominance. Papilio usually have a single locus governing polymorphic wing patterns, and morphs are determined by complete dominance in wing pattern inheritance6,19–21 whereas in Heliconius, several loci govern different aspects of wing patterning, and they often exhibit co-dominance among mimetic forms21,22. Complete dominance might have been selected in Papilio Batesian mimics to protect mimetic forms from maladaptive intermediates. On the other hand, selection of co-dominance in the aposematic Heliconius might have led to novel combinations of wing patterns, which appear to be favoured in this group23. To characterise dominance relationships in P. polytes, we performed a series of crosses between pure-breeding lines of all three female forms, designed to exploit the known female-limitation of mimicry and achiasmatic oogenesis (Fig. S3d). These crosses showed a clear and complete dominance hierarchy between the three P. polytes female form alleles, with romulus > polytes > cyrus, and all segregating broods showing ∼1:1 ratio of dominant and recessive phenotypes, as expected under complete dominance (Table S4–5). These dominance relationships, and the strict allelic inheritance and expression of mimicry in a sex-specific manner, were confirmed by genotyping of parents and female offspring using our form-specific primers (Methods, Table S2). None of the segregating broods showed intermediate or otherwise unusual wing colour phenotypes even when they were grown across different seasons in slightly variable climatic conditions and nutrition regimes, showing that environmental variation or genetic background did not affect the wing patterns (Table S4–5, personal observations). The female forms were thus controlled strictly by their allelic combinations under complete dominance.
The inferred sequence of origins of female forms and its inverse relationship with genetic dominance in P. polytes strictly follow the pattern predicted by Haldane: the ancestral, non-mimetic f. cyrus that evolved less than 9.58 mya is universally recessive, the next novel mimetic f. polytes=f. theseus that originated before 4.21 my is dominant over f. cyrus, and the most recent f. romulus that originated less than 1 mya is universally dominant (Fig. 1). These genetic dominance relationships and sex-limitation of mimicry do not break down in interspecific crosses with monomorphic or polymorphic sister species6 – contrary to the pattern observed in Heliconius numata where derived dominant alleles also show codominance24 – indicating that the genetic bases of these two critical adaptations are highly canalised, outweighing the potential impacts of different genomic backgrounds across species.
4. dsx shows signatures of selective sweeps and episodic selection, with rapidly evolving alleles
Having established that the dsx alleles underlie all the mimetic forms including the previously uncharacterised f. romulus, and that the successive mimetic forms in the P. polytes species group are sequentially dominant, we tested whether dsx shows signatures of selective sweeps and/or episodic selection as predicted under Haldane’s sieve. To put this in context, we characterised all the genomic regions that have experienced intense selection in this entire clade by estimating selective sweeps using Raised Accuracy in Sweep Detection (RAiSD). We found that dsx is the only gene that has experienced selective sweeps in all five mimetic species in this clade, but not in the basal monomorphic and non-mimetic relative, P. protenor (Fig. 2, Table S6. Each species had a different set of dsx SNPs showing signatures of selective sweeps (Fig. 2b). All these SNPs were in intronic regions, suggesting that there may be species-specific mutations associated with these SNPs that may have improved mimetic resemblance, which needs to be tested in the future. Further, the Branch-site Unrestricted Statistical Test for Episodic Diversification (BUSTED)25 revealed that dsx sequences show signatures of episodic diversifying positive selection in the mimetic species (p<0.0001), specifically in codons 142 and 148 (p<0.01) as revealed by Mixed Effects Model of Evolution (MEME)26.
Figure 2:
dsx is a hotspot of selective sweeps in all the mimetic species.
a. Annotated genes showing selective sweeps in the five mimetic species. The alternately coloured outer bands indicate chromosomes as defined in the reference Bombyx mori genome, whereas colour-coded lines inside represent genes showing signature of selective sweeps in the five species. b. Summary of analysis of selective sweeps using Raised Accuracy in Sweep Detection (RAiSD). Note that a single annotated gene may contain multiple loci (SNPs) that show signatures of selective sweeps.
How have the selective sweeps and episodic positive selection influenced molecular diversity and allelic differentiation in dsx in the context of mimicry? An analysis of single-nucleotide polymorphisms revealed that the three dsx alleles (HR, HP and h) were highly differentiated: pairwise comparisons of genome sequences of the three female forms revealed high average Fst (0.6 to 1) between variable loci of the three dsx alleles (Fig. S6). DNA sequences of HR, HP and h alleles (HR n=10; HP n=35; and h n=26) showed a total of 151 fixed substitutions (i.e., 13% of the exonic sequence), which have accumulated in short timeframes with the evolution of the three alleles (Fig. 3–4). Some of these substitutions are in the critically important domain regions of this conserved developmental master regulator, and a few involve amino acid substitutions (Fig. 3). For example, the OD2 domain of HR has some romulus-specific amino acid substitutions which might affect the protein-protein interactions of dsx that this domain facilitates (Fig. 3). Interestingly, all these allele-specific substitutions appear to fix early in the evolution of alleles, irrespective of how the female forms are parsed subsequently among species: the dsx gene tree showed a well-supported topology in which branches clustered first by colour forms and then by species, rather than the reverse—a pattern also seen in the haplotype network (Fig. S7). The dsx gene trees further corroborate the estimated evolutionary sequence of the origin of mimetic forms and the underlying dsx alleles (Fig. 1b–c). We did not find any species-specific fixed substitutions or polymorphisms in dsx coding sequences of the allelic variants.
Figure 3:
Allelic basis of mimetic polymorphism in the polytes species group.
Lepidopteran dsx comprises of six exons, of which exon 6 is untranslated27. It has female- and male-specific transcripts (called dsx F and dsx M, respectively), with multiple female isoforms (F1, F2 and F3) in the polytes group9,10. The exon composition of each isoform is depicted at the top. Allele-specific SNPs, their positions on the CDS and protein sequence, and their corresponding amino acids are colour coded. Invariable and non-specific polymorphic sites are represented by dotted lines. The domain regions (OD) are highlighted where feasible. OD2 spans several exonic regions in the DNA sequence where bases cannot easily be numbered because of different lengths of exonic regions across dsx isoforms. The amino acid sequence of OD1 is conserved.
Figure 4:
Rapid molecular evolution of mimetic dsx alleles in the polytes species group.
a. Allele-specific, fixed substitutions from Fig. 3 are shown for DNA and amino acid sequences (CDS) with respect to the evolutionary timeline of the origin of three dsx alleles (a linear increase is assumed for simplicity). The total number of fixed substitutions accumulated in each allele is shown before the new allele evolved. Colour-coded regions that cross evolutionary boundaries (dotted lines) represent the number of fixed substitutions that were inherited in the new allele from the previous allele from which it arose. For example, h has no fixed substitutions in the amino acid sequence but 16 in the DNA sequence, of which 7 were inherited in HP, of which 5 were inherited in HR. HP has 61 new, fixed DNA substitutions relative to h, of which 44 were inherited by HR. In addition, HR and h share 18 SNPs that are absent in HP, and 25 fixed substitutions that are unique to HR, showing a rapid accumulation of mutations in coding regions of novel mimetic alleles. See Fig. 3 for molecular details and sample sizes. b. Percentage of nucleotide substitutions in dsx sequence as observed at the genus and family levels (separated by a horizontal dotted line) for four insect orders 27. Numbers in parentheses after family/genus names represent the number of species from that group used in this analysis. The three alleles of dsx in P. polytes alone (vertical dotted line) have more substitutions than the dsx sequences within several genera.
The high molecular divergence between the coding regions of the three dsx alleles within the polytes species group, and especially between the mimetic HR and HP alleles of P. polytes alone in approx. 1 my, represents an exceptionally high rate of adaptive divergence under positive selection. Indeed, the percentage substitutions observed in dsx alleles within this single species is comparable to that across multiple insect genera (Fig. 4b). It is important to note, however, that it is unlikely that every substitution between HR and HP alleles is adaptive. It is possible that a considerable number of the fixed differences between HR and HP alleles might be a result of hitchhiking associated with strong selective sweeps caused by a few mutations of large fitness effects in the context of mimicry. In any case, this divergence may provide a specific case study and a benchmark for future comparisons of how insect dsx, although highly conserved across holometabolous orders, shows rapid differentiation in DNA sequence as well as protein structure when it is under selection for adaptive dimorphisms and polymorphisms in secondary sexual traits 27.
5. Rare exon swaps produce novel mimetic intermediates
The dsx alleles determining different female forms are protected by the inversion or lack of recombination, but how can novel forms evolve in this existing polymorphism? Do they arise strictly from existing forms in a sequential manner via accumulation of substitutions, as suggested above for existing female forms in the polytes species group? Or are there other mechanisms, such as evolution in isolation followed by hybridization and introgression28, or enhancer shuffling29, and differential developmental regulation30 by which new forms may be accommodated? A novel female form of P. polytes offers a window into this interesting problem. This intermediate form, which we first noticed in nature (Fig. S8, and was also observed by Clarke and Sheppard6) and then retrieved in a captive, mixed breeding population (Fig. 5a), had romulus-like forewings and polytes-like hindwings. We bred this form for over five generations, confirming that it was fertile and genetically stable. We examined the dsx sequence of two individuals from this pure-breeding line, which showed that they carried romulus-specific SNPs in exon 1, and polytes-specific SNPs in exons 2–5 (Fig. 5b–c). This implied that their ancestor had a recombination event in the approx. 35kb intronic region between exon 1 of HR and exon 2 of HP alleles. This intermediate phenotype demonstrates that romulus-specific SNPs in exon 1 (which contains the DNA-binding domain) may be sufficient to change the phenotype of the forewings but not produce a complete romulus phenotype, and that exons 2–5 of HP could override the dominance of HRallele to produce polytes-like hindwings. The protein-protein interaction domain that lies on other exons may be more critical for patterning hindwings, implying that dsx could have form-specific interacting partners, which is also suggested by the romulus-specific amino acid substitutions in OD2 domain of HR (Fig. 3). The 35kb region between exons 1 and 2 may also carry regulatory elements that affect the expression of dsx. Although, we do not observe an overall difference in dsx expression between these female forms (Fig. S5), the isoform dsxF1 seems to exclusively express in romulus females. The dsx alleles may thus manifest the dominant and recessive phenotypes using a combination of exonic regulation, differential isoform expression, novel downstream targets and interacting partners in each female form18.
Figure 5:
Exon swaps (recombination) between the HR and HP alleles produce a novel, rare intermediate phenotype.
a. Wing patterns and possible dsx genotypes of mimetic female forms of P. polytes, along with the two females with intermediate wing patterns that we obtained, are shown. Both the intermediates had a crossed over, hybrid dsx allele (represented as HH) with exon 1 of the universally dominant romulus allele and exons 2–5 of the second-dominant polytes allele. b. The three dsx alleles have distinctive SNPs in each exon, which are colour coded for the normal phenotypes that they produce. IBC-PT567 was heterozygous for polytes and cyrus alleles, while IBC-PT568 was homozygous for polytes allele, except that both specimens had exon 1 of HR allele. c and d. Exon-specific SNPs of romulus and polytes alleles in the intermediate IBC-PT567 and IBC-PT568, highlighted in blue. In panel c, romulus-specific SNPs are marked in red, and polytes-specific SNPs in green. Non-synonymous SNPs are highlighted in yellow.
This persistent intermediate phenotype suggests genetic means, e.g., a rare exon swap, by which novel mimicry phenotypes may be generated. This intermediate phenotype has no existing aposematic model to protect it from predation, so it may be considered at present to be maladaptive. However, such naturally occurring standing variation might become adaptive in other selective landscapes with different distasteful models. Interestingly, other wing pattern combinations, e.g., polytes-like forewings and romulus-like hindwings, or any other intermediate phenotypes, have never been observed in the field or in the lab. This suggests that the strong genetic architecture and selection together constrain the kind of standing genetic variation in wing patterns observed in this species group.
In light of our overall findings about the evolutionary history of mimetic polymorphism in the P. polytes species group and the novel female form just described, it is worth considering the roles of standing genetic variation versus new mutations, potential modification of dominance relationships in relation to Haldane’s sieve, and maintenance of genetic mimetic polymorphisms under long-term balancing selection. There was substantial discussion and speculation about these aspects from the time of the Modern Synthesis to early breeding experiments to probe the inheritance of mimicry31–33. With theoretical and molecular genetic advances, a much more nuanced hypothesis has developed. The consensus seemed to be that for Batesian mimicry to evolve, an initial mutation would be required that establishes partial mimetic resemblance to an aposematic model. Due to the complexity associated with wing pattern phenotypes, it is likely that additional subsequent mutations would be necessary to improve mimetic resemblance, and these were termed “modifiers”34,35. A modifier could add to mimetic resemblance itself, make it partially or completely dominant, and/or reduce recombination to ensure that the mutations associated with mimicry did not break down and were inherited together. Therefore, modifiers could encompass an array of mutations ranging from point mutations that alter gene function to large inversions and rearrangements that could give rise to supergene architecture. The mimicry “gene” could subsequently either go to fixation, or the mimetic and non-mimetic allelic combinations could be maintained by balancing selection in polymorphic species under negative frequency dependence. In Papilio polytes, it was assumed that mimetic polymorphism is maintained by long-term balancing selection on the underlying allelic variation at the mimicry gene dsx, as HKA tests showed high genetic diversity at this locus10,34. Our work defines the timescales of these long-term balancing selection events to be up to 9.58 my and 1 my for the mimetic forms in the P. polytes species group (Fig. 1 and 4, Section 1 of Results and Discussion). Additionally, our analysis revealed that dsx has experienced selective sweeps in the mimetic species of the P. polytes species group but not in the basal non-mimetic P. protenor, implying that mimetic alleles experienced a rapid increase in frequency in the population. This contrast of high genetic diversity and signatures of sweep could co-occur if the mimetic allele shared among the species swept through the population under a soft sweep between 9.58 my and 4.21 mya, before the mimetic species split from each other. The selective sweeps likely caused moderate levels of genetic hitchhiking, and the alleles subsequently accumulated mutations due to lack of recombination and rapid molecular evolution at this locus11,12, resulting in high genetic diversity at the locus with some remnants of a sweep.
The origin of the first mutation causing mimetic resemblance may affect the spread of this trait through the population. While this may have come from existing standing genetic variation or new mutations, the latter has largely been implicated in mimicry 31–33. The dsx gene lies within a specific inversion that seems to be unique to the P. polytes species group as we have found no matching inversion breakpoint sequences outside of the polytes species group. Yet, dsx seems to have been repeatedly recruited in mimetic resemblance in other closely related groups as well, however, with other unique allelic combinations and independent smaller inversions16,17. Therefore, along with our genetic analysis of allelic variations and selective sweeps, current evidence appears to support Fisher, Sheppard and Ford’s speculations that mimicry in the P. polytes species group may have evolved in large part from novel mutations at dsx, many of which apparently sweeping through the populations, rather than from standing genetic variation that was shared between polytes and memnon species groups. Under this scenario, the predictions of Haldane’s sieve would be valid and directly determined by the dominance of new mutations36.
The more challenging aspect of Haldane’s sieve is to identify whether a novel mutation contributing to mimetic resemblance is partially or completely dominant at the point of origin, or it may subsequently become dominant under selection. Under Batesian and Müllerian mimicry, evolution of dominance modifiers may allow an initial recessive mutation to become partially or completely dominant, thereby facilitating its spread through the population37–39. It is currently unclear what may constitute a dominance modifier in the context of mimetic resemblance, for example, a regulatory element may alter the expression levels of an allele or an epistatic interaction may alter the dominance of a mutation40. However, in terms of evolutionary dynamics under Haldane’s sieve, the spread and the subsequent evolution of this mimicry mutation would be impacted by the point of dominance, not the origin. It is currently methodologically challenging to deduce whether dominance arose with the first mutation leading to a partial mimetic resemblance or evolved subsequently as mimicry was perfected with additional mutations31,32, and how much fitness benefit successive mutations offered41. In the future, a combination of genetic and developmental manipulations may perhaps be able to throw light on these aspects.
Our recent finding that P. polytes is a complex of three species13 makes it necessary to reevaluate previous genetic studies that assumed javanus and alphenor to be subspecies of P. polytes. Clarke and Sheppard’s characterisation of inheritance of the female forms was based on interspecific hybrids between the three species rather than segregating broods of the same species6. The first genome-wide mapping and developmental expression studies of mimicry polymorphism were done in the Philippine P. alphenor, not in P. polytes10. Subsequent reference genome and developmental studies found inversion breakpoints and differential expression of dsx largely in the Japanese populations of P. polytes9,42. Previous studies determined the evolutionary history of mimetic forms in this group based on samples from different species, which has possibly led to incorrect conclusions regarding the origin of f. theseus12. However, conclusions regarding the developmental genetic basis of mimicry from the present and previous genetic studies on P. polytes and P. alphenor are not contradictory, showing that: (a) the developmental genetic basis of mimicry and female polymorphism is shared between both the species, and (b) dsx controls mimetic polymorphism by means of: (i) an inversion—which predates the diversification of all mimetic species in the group—that isolates mimetic from non-mimetic alleles, (ii) allelic variation and differential gene expression across males/f. cyrus and mimetic females, producing sex-limitation of the mimetic polymorphism, and (iii) tissue-specific isoform expression across developmental stages to accommodate novel adult polymorphisms that are regulated in late development9,10,42.
Our work contains extensive sampling of the polytes species group, including dozens of specimens of all known female forms from mainland Asia, specifically South Asia where mimetic polymorphism is most striking, and several island groups that were not sampled in previous studies. This extensive sampling and our reconstruction of the evolutionary history of mimetic polymorphism in this group, the origin of each female form, the dominance relationships between alleles of dsx, and molecular evolution dsx in relation to mimetic polymorphism, provided unparalleled insights into the evolutionary and genomic history of this entire species group that substantially contrast with previous studies6,12,13. We have characterised allele-specific mutations across the dsx gene, which could be used for functional assessment and identification of key mutations that affect mimetic phenotypes. We demonstrate that the female forms in the polytes species group have evolved in a step-wise fashion through Haldane’s sieve— each successive novel mimetic form having complete dominance over pre-existing forms. Further, we find that novel wing phenotypes could arise from rearrangements in existing form-specific alleles, through exon swaps, and that different exons of the same gene could regulate colour patterns on the forewing and hindwing. Our work also sheds light on how dominance may interact with other genetic and population genetic phenomena, such as recombination and selective sweeps, to influence evolutionary trajectories of diversification of lifeforms.
Specimen collection, genome re-sequencing and SNP calling
We preserved wild-caught P. polytes and P. protenor in 100% ethanol across the species range (Fig. S1, Table S1. We also obtained P. alphenor and P. javanus from native commercial butterfly breeding facilities in the Philippines and Java, respectively, and preserved them in 100% ethanol. Sample details are in Table S1. We extracted DNA from thoracic muscle of preserved samples using QIAGEN DNeasy blood and tissue kit, quantified the extracted DNA using Qubit fluorometric quantification, and prepared libraries using Illumina TruSeq DNA PCR-free library preparation kit. We used a 2×100 PE run on Illumina HiSeq 2500 for re-sequencing genomes of our Papilio samples. We also downloaded available genome sequences from the SRA database for P. alphenor, P. javanus, P. phestus, P. ambrax and P. protenor. We checked the quality of raw reads, aligned them to the Papilio polytes reference genome (Ppol_1.0, genome version 1.0, annotation version 1.0) from NCBI9, using BWA aligner43, and called SNPs using the GATK pipeline44. We processed the alignment files to mark and remove duplicates, merge files from the same sample, performed indel realignment, called haplotypes, and combined the resulting VCF files for genotype calling. We filtered the resulting SNPs using the recommended parameters in the GATK manual. The resulting dataset contained samples that had an average coverage per sample of 9X across the genome; and 13X and 8X across the mimetic and non-mimetic doublesex scaffolds respectively. Exons of the dsx gene showed nearly complete coverage and higher depth compared to introns. Post filtering of called SNPs, we obtained a set of 27,965,662 SNPs for the polytes species group. We extracted dsx CDS from individual alignments using bcftools-1.6 mpileup45. We aligned exons manually and annotated allele-specific SNPs to generate the SNP map for dsx in Extended Fig. 3. We identified sites that were fixed in all samples of each female form to generate consensus CDS for each dsx allele. We also sequenced genomes of two intermediate female individuals from a pure-breeding line, aligned and processed them as given above, and manually annotated SNPs to find out their exonic composition (Fig. 5). We performed nucleotide and protein alignments in MEGA X46 and visualised them in Jalview47.
Reconstruction of species phylogeny, gene trees, and haplotype network
We extracted CDS and non-coding sequence of dsx from the genome re-sequencing data using bcftools-1.6 mpileup45. We aligned sequences with Muscle aligner48 in MEGA X, using codon alignment for the dsx coding sequence. We used PartitionFinder 2 to find appropriate models of evolution for each set of sequences: F81+G, GTR+I and JC for the dsx CDS dataset, GTR+I+G for the dsx non-coding dataset. We reconstructed dsx gene trees using MrBayes 3.2.7a with the same parameters as mentioned above. We viewed all phylogenies using FigTree v1.4.349. We filled gaps in the CDS extracted from individual alignments using exon-specific PCRs, followed by Sanger sequencing. Primer details are provided in Table S2. We used popart-1.750 to generate a haplotype network of dsx alleles using TCS method (Fig. S7).
Estimation of divergence times
We acquired sequence data for four mitochondrial markers (cytochrome c oxidase subunit-I, tRNA leucine, cytochrome c oxidase subunit-II and 16S) and two nuclear markers (elongation factor-I alpha and wingless) from a broad range of Papilionidae species (Condamine et al 202315 and Joshi and Kunte Biorxiv51) to estimate the time of origin of the mimics and models. We aligned the sequences using MAFFT v7.47552 and visually verified the accuracy of the alignments using MEGA X46. We used PartitionFinder 2.1.153 to identify the best-fitting partitioning scheme and corresponding models of evolution. We used BEAST v2.7.454 for generating the time-calibrated phylogeny. We applied an optimized relaxed clock model and a birth-death model as molecular clock and tree priors, respectively. For time calibration, we used three primary calibration points (the crown ages of (1) Papilionidae, between 47.80 and 150 Ma, (2) Parnassiinae, between 23.03 and 150 Ma, and (3) Luehdorfiini, between 5.33 and 150 Ma) and 10 secondary calibration points (the crown ages of (1) Papilioninae, between 34.40 and 62.90 Ma, (2) Leptocircini, between 26.60 and 49.90 Ma, (3) Troidini, between 26.90 and 50.40 Ma, (4) Papilionini, between 27.50 and 50.90 Ma, (5) Old World Papilio, between 21.52 and 33.5 Ma, (6) subgenus Heraclides, between 16.07 and 27.48 Ma, (7) subgenus Araminta, between 9.89 and 18.61 Ma, (8) subgenus Papilio, between 7.01 and 13.85 Ma, (9) subgenera Achillides, Princeps, and Menelaides, between 15.57 and 24.39 Ma, and (10) subgenus Menelaides, between 10.90 and 17.65 Ma) with uniform priors15. We ran six independent MCMC chains for 150 million generations and sampled every 15,000 generation. We performed the phylogenetic analyses on the CIPRES web server ( We combined the results using LogCombiner after removing 25% of the samples as burnin and ensured convergence by evaluating effective sample sizes of relevant parameters on Tracer v1.7.155. We constructed the maximum clade credibility (MCC) tree using TreeAnnotator after removing 25% of the samples as burnin. Divergence times estimated by our work were similar to those estimated in previous studies, although we included many more Menelaides samples and additional outgroups to estimate node ages of models and mimics in these mimicry relationships.
Estimation of signatures of selection in P. polytes, P. javanus and P. alphenor
To identify loci showing signatures of selection in the genomic dataset, we filtered the SNP dataset (total 27 million SNPs) by species and calculated mu statistic using Raised Accuracy in Sweep Detection (RAiSD) v.2.556. We used a conservative cutoff (>99.5%) of the µ statistic to identify loci experiencing selective sweeps. To minimise the possibility of false positives, we randomly resampled SNPs from a subset of samples to calculate the false positive rate threshold in RAiSD. The resulting threshold was lower than the 99.5% cutoff we had used, so it did not eliminate any resulting hits. The number of identified SNPs experiencing selective sweeps that were included within annotated genetic elements and those that were in the intergenic regions lacking annotation features are given elsewhere13. The identities of annotated loci experiencing selective sweeps among the species are listed in Table S6. We mapped the genes experiencing selective sweeps in each species and common across the five mimetic species to the Bombyx mori genome57 using BLAST+ to identify their chromosomal locations. We used Circos-0.69-958 to map these genes across chromosomes for each species, assuming synteny between P. polytes and Bombyx mori genomes.
We used MEME26 to estimate the sites subjected to episodic positive or diversifying selection (Fig. 2) between dsx alleles. We used BUSTED25 to test for positive selection by asking whether dsx has experienced positive selection in at least one site in the allelic variants. We implemented MEME and BUSTED in HyPhy 2.559 using our dsx gene tree of coding sequences (Fig. S7).
Cross design and dominance hierarchy
We used mated wild-caught females from NCBS campus to start P. polytes greenhouse populations, which we maintained in large (2x2x2m) cages. We raised larval broods at approx. 28±4°C, on a diet of lemon (Citrus sp.) and curry plants (Murraya koenigii) and maintained the adults on Birds Choice butterfly nectar. We generated mapping broods (design described in Fig. S3d) by separating mating pairs from the population and letting the mated females lay eggs in individual small (0.6×0.6x1m) cages. Because of achiasmatic oogenesis, we then back-crossed the heterozygous male offspring normally with homozygous recessive females from the greenhouse populations. For GWAS, we generated a total of six segregating broods (1:1 mimetic:non-mimetic female progeny), three each segregating for polytes/cyrus and romulus/cyrus phenotypes. Among these, we sequenced the parents and 50 female offspring of each form from a romulus-cyrus segregating brood that produced 426 offspring. We used these six broods as well as similarly generated romulus-polytes segregating broods for genotyping with form-specific primers (Table S2) to establish the dominance hierarchy (TableS4–5). Among 290 individuals genotyped from 10 mapping broods, we observed no cross-reactivity of primers between forms, indicating that these SNPs are indeed form-specific, and that the colour forms are inherited and expressed in a sex-specific manner.
Genome-wide association study
We used a reduced representation genotyping-by-sequencing (GBS) approach to sequence offspring of the largest romulus-cyrus segregating brood, with a standardised protocol for generating a marker set using PstI and MluCI–mediated shearing of DNA, followed by library preparation and sequencing on an Illumina platform using 75 bp SE module (Genotypic Technologies Pvt. Ltd.). We used a TASSEL GBS pipeline with the Papilio polytes reference genome for SNP calling60 and tested the association of SNPs with female forms using a dominant model. We found very few reads from romulus individuals mapping to the non-mimetic cyrus allele of dsx from the reference genome. This may be due to low representation and coverage of the dsx gene with the GBS method, or due to mismatch between the highly divergent romulus and cyrus dsx sequences. We used Bonferroni correction to account for multiple comparisons to call significant associations. We used BLAST on NCBI and KaikoBase platforms to annotate the sites that associated significantly with f. romulus (Fig. S3).
Testing genetic divergence between the dsx alleles
To find genetic divergence between dsx sequences from different samples, we grouped our dataset by female forms, and extracted SNP data for the dsx mimetic and non-mimetic scaffolds. We calculated Weir-Cockerham’s Fst for the three female forms using vcftools 0.1.1361 (Fig. S6). To compare substitution rates between dsx alleles in P. polytes and other Lepidoptera, we used dsx alignments from Baral et. al., 201927, and identified substitutions in dsx sequences at the level of genera and families (Fig. 4b).
Breakpoint characterization for f. romulus
To identify potential breakpoints of HR, we designed primers flanking breakpoints of HP 9. We performed PCRs with lab-bred and wild-caught samples of all three female forms, cleaned the PCR products with ExoSAP-IT cleanup reagent and sequenced them using Sanger sequencing. We then manually aligned the sequences of the left and right breakpoints in MEGA X and annotated the alignment using Jalview (Fig. S4).
qPCR for dsx expression
We extracted RNA from 3-day pupal tissue using phenol-chloroform extraction method. We synthesized cDNA using ProtoScript II First Strand cDNA Synthesis Kit (New England Biolabs) and performed qPCR with existing primers for dsx and its isoforms9 using RPL3 as an endogenous control (our RPL3 primers given in Table S2, Fig. S5).
Images are for reference only.Images and contents gathered automatic from google or 3rd party sources.All rights on the images and contents are with their legal original owners.
Aggregated From –
Comments are closed.